Epitranscriptomics and the emerging roles of
RNA modifications in RNA function
While “GENE” and “GENE” both spell the same, formatting of the latter word changes the emphasis we place on it. This is analogous to the study of epitranscriptomics, where the underlying RNA sequence can be the same but chemical RNA modifications (epitranscriptome) change how the cell regulates and expresses the RNA. This is why epitranscriptomics research has revealed new regulatory pathways that could not be determined from simply sequencing the transcriptome. To date, research has demonstrated that the epitranscriptome regulates almost all forms of known RNA processing pathways. Consequently, changes in the epitranscriptome or misregulation of epitranscriptomic factors contribute to the progression of a multitude of diseases. Therapeutics that target epitranscriptomic factors, which include “writers” and “erasers” that respectively add or remove the N6-methyladenosine (m6A) modification as well as “readers” that target m6A, are already scheduled for clinical trials in 2021.
The goal of our laboratory is to discover new RNA modifications and epitranscriptomic factors as well as determine their mechanisms for regulating molecular and cellular processes and how these ultimately impact the progression of various diseases. We do this by answering the WHERE, the WHAT, & the WHY of RNA modifications.
ONGOING PROJECTS:
1) Develop technologies for high-resolution epitranscriptome sequencing to map WHERE RNA modifications are

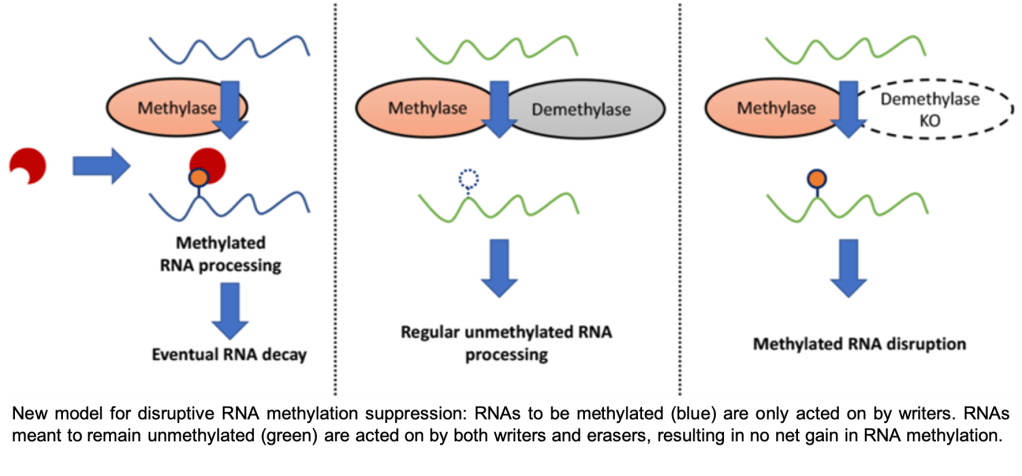
The invention of single-base-resolution RNA modification (epitranscriptome) sequencing has revealed great insights into epitranscriptomic regulation of cellular processes. For instance, our lab developed m6A-crosslinking-exonuclease (m6ACE) sequencing for quantitative single-base-resolution mapping of transcriptome-wide RNA methylation profiles. Using m6ACE, we generated the first ever human atlas of RNA methylomes distinct to each and every RNA writer or eraser known thus far. Insights garnered from this atlas allowed us to challenge the notion that erasers mediate cycles of methylation reversal and furthermore, thereby redefining their functional roles as suppressors of aberrant RNA methylation (Koh et al. 2019). Therefore, this highlights the importance of resolution in epitranscriptome sequencing.
Given that there are many other RNA modifications that still lack a precise mapping method, we are interested in developing more tools to sequence other RNA modifications of interest at precise resolution.
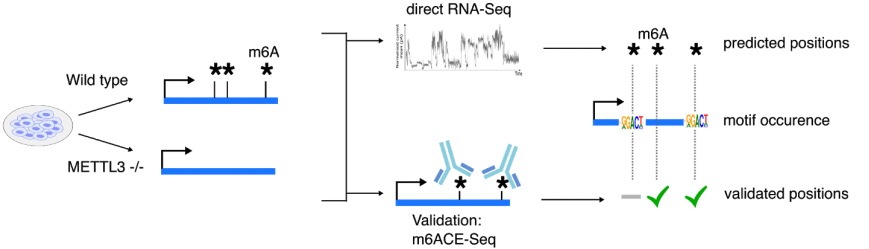
Another form of high-resolution sequencing is single-cell-resolution. Just as how single-cell transcriptome-sequencing has revealed new cellular subtypes that are stratified by their gene expression signatures, mapping single-cell epitranscriptomes can likewise reveal new cellular subtypes that are stratified by specific RNA modifications. We are currently exploring techniques that will facilitate single-cell epitranscriptome sequencing. For instance, we have recently explored antibody-independent methods for sequencing RNA modifications. By harnessing the quantitative precision of m6ACE, we generated training datasets for developing a new algorithm, xPore to map and quantify m6A methylation using direct RNA sequencing, generating transcriptome-wide and single-base-resolution RNA methylomes (Pratanwanich et al. 2020).
Relevant publications:
Goh WSS. RNA modifications: one ring to map them all (2026) Nature Reviews Molecular Cell Biology.
Pratanwanich PN* et al. Goh WSS*, Göke J*. Identification of differential RNA modifications from nanopore direct RNA sequencing with xPore (2021) Nature Biotechnology.
Koh CWQ, Goh YT, Goh WSS*. Atlas of quantitative single-base-resolution N6-methyl-adenine methylomes (2019) Nature Communications.
Koh CWQ et al. Goh WSS*. Single-nucleotide-resolution sequencing of human N6-methyldeoxyadenosine reveals strand-asymmetric clusters associated with SSBP1 on mitochondrial genome (2018) Nucleic Acids Research.
2) Identify and functionally characterize novel epitranscriptomic factors to determine WHAT RNA modifications do
RNA is modified by catalytic Writers that often function in a complex with co-factors guides, which direct Writers to modify specific RNA sequences. Identification of these Writers and co-factors is required for generating the knockout (KO) cell lines that form the basis of necessary tools for studying the function of the associated RNA modification. In our recent work, we screened for candidate RNA methylation Writers and used m6ACE to demonstrate that Mettl4 is a novel N6-methyl-2’O-methyl-adenosine (m6Am) Writer. By generating METTL4-KO cells, we demonstrated that Mettl4 absence causes loss of methylation on U2 snRNA, leading to a global change in pre-mRNA splicing patterns (Goh et al. 2020). Following up on this, we will explore other novel epitranscriptomic Writer/co-factor candidates and characterize the function of their associated RNA modification.

We were also one of the first groups to identify PCIF1 as a novel writer for TSS-m6Am by using m6ACE to map differential RNA methylomes between wildtype and PCIF1-KO cells at the TSS (Koh et al. 2019).

Unlike its well-characterized cousin m6A, there was no consensus on m6Am’s functional role. We hypothesized that by finding the protein factors (Readers) that recognize m6Am and knowing what the Reader does, this will reveal how m6Am truly functions. To that end, we identified transcriptional terminator PCF11 as a novel m6Am Reader, revealing an unexpected role for m6Am in suppressing premature transcriptional termination and driving selective gene expression. Mechanistically, m6Am sequesters PCF11 away from RNA Pol II, thereby suppressing PCF11 from terminating RNA Pol II transcription prematurely (An et al. 2024).

We are currently using biochemical and omics tools to identify and mechanistically characterize other novel epitranscriptomic factors.
Relevant publications:
An HH, et al. Goh WSS*. m6Am sequesters PCF11 to suppress premature termination and drive neuroblastoma differentiation (2024) Molecular Cell.
Goh YT, et al. Roca X*, Goh WSS*. METTL4 catalyzes N6-methylation of adenosines in U2 snRNA to regulate pre-mRNA splicing (2020) Nucleic Acids Research.
Koh CWQ, Goh YT, Goh WSS*. Atlas of quantitative single-base-resolution N6-methyl-adenine methylomes (2019) Nature Communications.
3) Explain WHY RNA modifications are important by investigating the roles of epitransciptomics in disease and therapy
We aim to utilize the technolgies we developed to map WHERE RNA modifications are, combined with the discoveries we made for WHAT RNA modifications do, to answer WHY RNA modifications are important and what their impact in diseases are. For instance, high-risk neuroblastoma is characterized by overamplification of the MYCN gene, and is treated using all-trans-retinoic-acid (ATRA). Using m6ACE’s quantitative precision, we found that ATRA induces m6Am modification of ATF3, which is in turn a repressor of MYCN expression. Given that we had determined m6Am to sequester/suppress PCF11 to promote expression of m6Am genes, we were able to uncover m6Am’s vital role in contributing to ATRA’s therapeutic efficacy in driving neuroblastoma cell differentiation and suppressing their tumour stem-like properties (An et al. 2024).
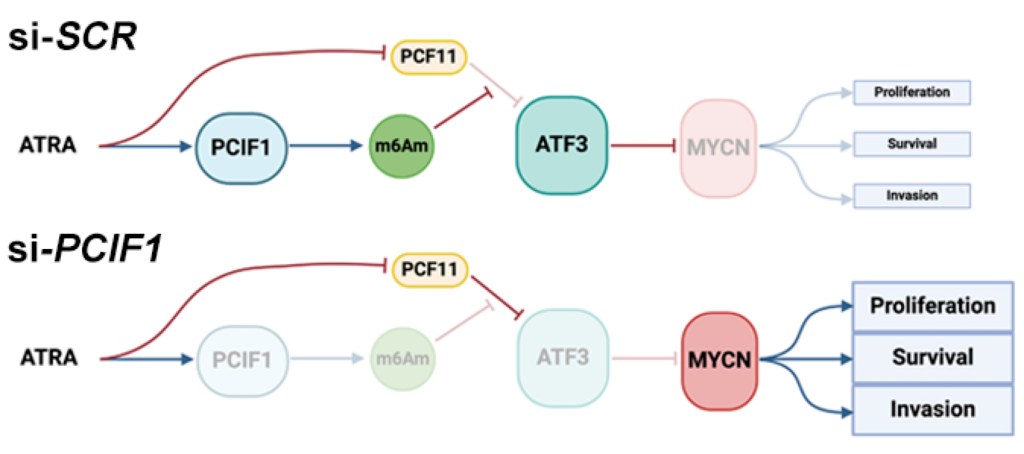
We will eventually incorporate the other novel sequencing technologies that we are developing together with novel epitranscriptomic factors that we are characterizing so as to expand this research angle to investigate the role of epitranscriptomics in other diseases.
Relevant publications:
An HH, et al. Goh WSS*. m6Am sequesters PCF11 to suppress premature termination and drive neuroblastoma differentiation (2024) Molecular Cell.
